mRNA-Seq
CD Genomics's mRNA-Seq service, powered by the state-of-the-art Illumina NovaSeq platforms, offers comprehensive solutions for gene expression quantification, differential gene expression analysis, identification of novel transcript isoforms, alternative splicing, and gene fusions, etc. Our expert team works closely with you to provide standard and customized high-throughput sequencing and bioinformatics analyses based on project-specific needs, as well as exquisite results for publication.
Overview
Messenger RNA (mRNA) is one type of RNA transcripts that corresponds to the genetic sequence of a gene, instructing the protein synthesis in cells. Eukaryotic mRNA sequencing (mRNA-Seq), by leveraging the technology of next-generation sequencing (NGS), reveals the expression profiles of mRNA in a given biological sample, as well as differential gene expression among sample groups. The mRNA-seq provides a clear, complete, high-resolution view of the coding transcriptome, including detection of known and novel transcript isoforms, identification of gene fusion events and other features, as well as gene expression analysis by measuring transcript abundance. In contrast with whole transcriptome sequencing that sequences all coding and non-coding transcripts, mRNA-seq focuses on the coding RNAs, which are around 2% of the whole transcriptome and contain a poly-A tail. The mRNA-seq delivers a comprehensive picture of the coding transcriptome without the need of prior knowledge and is advantageous over gene expression arrays and other approaches. mRNA-seq has been widely used for identification and comparison of gene expression levels between groups, perfect for drug treatment, behavioral selection, identification of cellular responses to viruses, and disease related research.
Service Portfolio
Eukaryotic De Novo mRNA Sequencing
De novo sequencing directly sequences the genome or transcriptome without any sequence information as a reference. Our eukaryotic de novo sequencing utilizes the Illumina platform to sequence all mRNAs transcribed by a specific eukaryotic tissue or cell in a specific state, and spliced out the longest transcript as unigene for subsequent analysis.
Eukaryotic mRNA Sequencing with Reference
Eukaryotic mRNA sequencing based on reference sequences can comprehensively obtain transcript information of species-specific cells, tissues, or organs, so as to conduct eukaryotic transcript structure analysis, variation analysis, gene expression level analysis, novel transcript discovery, and more.
Nonsense-Mediated mRNA Decay (NMD) Analysis
Nonsense-mediated mRNA decay (NMD) is a mRNA quality monitoring mechanism that exists widely in eukaryotic cells. CD Genomics provides NMD analysis services for further elucidation of the mechanism of gene expression.
Prokaryotic mRNA Sequencing
CD Genomics prokaryotic mRNA sequencing service is based on the Illumina high-throughput sequencing platform, capable of detecting the overall transcript level of any species at the single nucleotide level.
Blood RNA Sequencing
Our comprehensive blood RNA sequencing services to characterize total RNA in blood samples, including, circle RNA, mRNA and small RNA. Our advanced whole blood transcriptional profiling solution ensures reproducible, specific, sensitive and accurate results to aid disease research and biomarker discovery.
Features
| Flexibility | Sensitivity | Optimization | Application |
|---|---|---|---|
| Flexibility to prepare libraries and analyze data according to your needs. | Enables sensitive and accurate isoform detection and quantification. | ptimized workflows to maximize output and accuracy. | Especially useful for medical research and agricultural research |
Project Workflow

1. Sample Preparation
RNA purification; quality assessment and quantification
2. Library Preparation
cDNA library or strand-specific library

3. Sequencing
Illumina NovaSeq, PE 150

4. Data Analysis
Visualize and preprocess results, and perform custom bioinformatics analysis.

Bioinformatics Analysis Pipeline
In-depth data analysis:
- Data Quality Control
- De novo assembly or reference assembly
- Gene (including oncogene) functional annotation
- Gene expression quantification
- Differential expression profiling
- GO and KEGG enrichment analyses
- Protein-Protein Interaction (PPI) analysis
- Transcription factors prediction
- SNP & InDel detection
- Alternative splicing analysis
- Fusion gene prediction
Sample Requirements
- RNA sample (quantity ≥ 1 μg, concentration ≥ 300 ng/μL)
- RIN ≥ 6.8 with smooth base line; 28S/18S ≥ 1.5; OD260/280 =1.8-2.2; OD260/230: ≥ 1.8.
Sample storage: RNA can be dissolved in ethanol or RNA-free ultra-pure water and stored at -80°C. RNA should avoid repeated freezing and thawing.
Shipping Method: When shipping RNA samples, the RNA sample is stored in a 1.5 mL Eppendorf tube, sealed with sealing film. Shipments are generally recommended to contain 5-10 pounds of dry ice per 24 hours.
Deliverable: raw data as BAM files, coverage summary, QC report, custom bioinformatics analyses.
Demo Results
 Transcriptome Alignment Analysis Results
Transcriptome Alignment Analysis Results
 Differential expression of known genes
Differential expression of known genes
 Differential expression profiling
Differential expression profiling
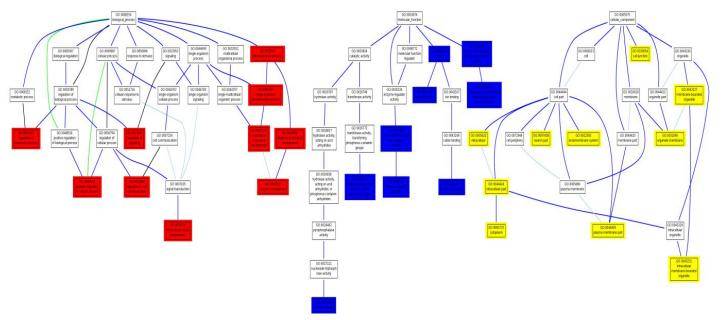 GO enrichment
GO enrichment
FAQ
-
- What are the requirements for blood samples to do RNA-seq?
-
- What is the recommended data volume for eukaryotic mRNA-seq?
-
- Why is mRNA sequencing recommended compared to microarray expression profiling?
-
- What are FPKM and RPKM?
References:
- Wahlstedt H, Daniel C, Ensterö M, et al. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome research, 2009, 19(6): 978-986.
- Park B J, Ahn H S, Han S H, et al. Analysis of the Immune Responses in the Ileum of Gnotobiotic Pigs Infected with the Recombinant GII. p12_GII. 3 Human Norovirus by mRNA Sequencing. Viruses, 2021, 13(1): 92.











