Comprehensive Guide to RNA Extraction: From Principles to Practical Applications
Innovate with Us





Ribonucleic acid (RNA) plays a central role in various biological processes, including the transmission of genetic information, protein synthesis, and cellular metabolism regulation. Serving as the molecular messenger between deoxyribonucleic acid (DNA) and proteins, RNA participates in the precise regulation of cellular activities through multiple mechanisms, such as transcriptional regulation, translational guidance, and epigenetic modifications.
In molecular biology research, RNA extraction is a fundamental technique essential for conducting transcriptome analyses, investigating gene functionalities, and exploring disease mechanisms. Through technologies such as quantitative real-time polymerase chain reaction (qRT-PCR), digital PCR (dPCR), microarray analysis, and high-throughput RNA sequencing (RNA-seq), researchers can achieve accurate quantification of specific gene expression levels and comprehensive transcriptome profiling based on high-quality RNA samples. This, in turn, allows for the elucidation of dynamic changes in gene expression regulatory networks under pathological and physiological conditions.
This article focuses on the standardized operational procedures for RNA extraction and underscores its critical role in molecular biology research.
Comparative Analysis of Common RNA Extraction Methods
1 Principles and Technical Advantages of the Trizol Method
The Trizol method is a classic technique for RNA extraction, based on the synergistic action of guanidine isothiocyanate and phenol. Guanidine isothiocyanate acts as a potent denaturing agent, rapidly disrupting the cellular structure and leading to the denaturation of proteins within the nuclear protein complex, thereby releasing RNA. Phenol further aids in cell lysis and plays a critical role in the subsequent phase separation process.
Under the influence of Trizol reagent, the cellular lysate undergoes a series of treatments that result in phase separation. Specifically, upon the addition of chloroform and subsequent centrifugation, the solution forms three distinct layers: the bottom layer, consisting of the phenol-chloroform phase, primarily contains hydrophobic proteins and DNA; the middle layer is the denatured layer, which contains cellular debris and other impurities; the top aqueous phase is enriched with RNA. This principle of phase separation effectively allows for the isolation of RNA from other biomolecules, providing a foundation for subsequent purification.
The technical advantages of the Trizol method include:
- Broad sample applicability, suitable for various biological samples such as animal tissues, plant materials, and microorganisms.
- Standardized operation procedure that does not rely on specialized instruments, enabling even novice researchers to perform the protocol correctly.
- Excellent extraction efficiency, allowing for the acquisition of high-integrity RNA (RIN ≥ 8), with an A260/A280 ratio between 1.8 and 2.0, and acceptable protein residue (A260/A230 > 2.0) and genomic DNA contamination levels, all of which meet the requirements for molecular experiments within 30 minutes.
However, it is important to note the limitations of this method: the presence of phenolic compounds and guanidine isothiocyanate poses cytotoxicity, necessitating the execution of the procedure within a fume hood and adherence to strict personal protective measures.
Learn More: TRIzol RNA Extraction Protocol
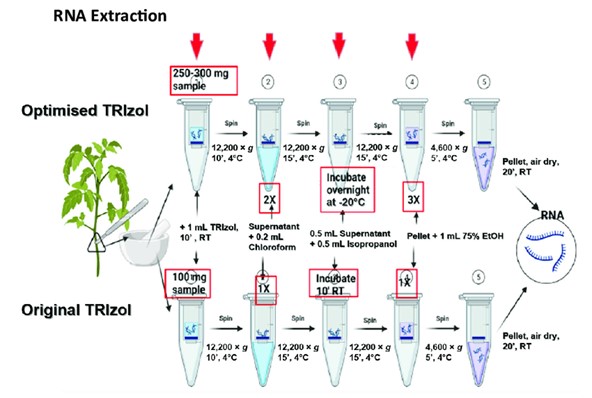 Workflow of RNA extraction using TRIzol reagent (Anis Afifah et al,. 2021)
Workflow of RNA extraction using TRIzol reagent (Anis Afifah et al,. 2021)
2. Guanidine Hydrochloride and Lithium Chloride Methods
In addition to the Trizol method, the guanidine hydrochloride-organic solvent method and the lithium chloride-urea method are also commonly employed for RNA extraction, each possessing distinct characteristics.
The guanidine hydrochloride-organic solvent method inhibits RNase activity through the application of guanidine hydrochloride and utilizes organic phase extraction to remove proteins. However, this method entails a relatively complex series of steps and is time-consuming, resulting in a lower extraction efficiency compared to other techniques.
Conversely, the lithium chloride-urea method employs high concentrations of urea to denature proteins, followed by the selective precipitation of RNA using LiCl. This approach is quicker and more straightforward, yet it has notable drawbacks, such as the propensity for the loss of small RNA molecules like 5S RNA, as well as a heightened risk of genomic DNA contamination, with the A260/A280 ratio often falling below 1.8.
The limitations of both methods become particularly pronounced when dealing with specialized samples, such as plant tissues rich in polysaccharides and polyphenols. In these cases, researchers must exercise caution in their selection. While the lithium chloride-urea method may be considered a viable option for those prioritizing speed, it is crucial to be aware of the associated contamination risks. On the other hand, although the guanidine hydrochloride-organic solvent method is more complex, it may be adapted for use in experiments requiring high RNA integrity through optimization.
3 Silica Gel Membrane Adsorption Method
With the advancement of biotechnology, the silica gel membrane adsorption method has gained widespread application in the field of RNA extraction, particularly in commercial reagent kits. This method offers a rapid purification process and an automated workflow, providing researchers with an efficient and convenient alternative for RNA isolation.
The primary mechanism of the silica gel membrane adsorption method relies on the specific affinity of silica gel membranes for RNA molecules. Under defined buffer conditions, RNA can tightly bind to the silica gel membrane, while other contaminants are removed with the elution buffer, thereby facilitating rapid purification. This approach significantly reduces extraction time and enhances experimental efficiency.
Numerous commercial reagent kits integrate automated devices with the silica gel membrane adsorption method, further streamlining the operational workflow. Researchers are required only to introduce samples into the kit and follow a predetermined protocol, after which the device autonomously executes a series of steps, including cell lysis, adsorption, washing, and elution. This automation minimizes human error and improves experimental reproducibility.
Nevertheless, the silica gel membrane adsorption method encounters a challenge concerning the potential residual DNA contamination. To effectively mitigate DNA residuals, it is advisable to incorporate an appropriate amount of DNase I during the extraction process for digestion. Furthermore, optimizing the washing steps—by either increasing the number of washes or adjusting the composition of the washing buffer—can contribute to reducing DNA contamination, thereby ensuring that the purity and quality of the extracted RNA meet the requirements for subsequent experimental applications.
RNA Sequencing Sample Submission and Preparation Guidelines and A Beginner's Guide to RNA Sequencing may be useful articles for your RNA sequencing projects.
Standardized Operating Procedure for the Trizol Method
1. Sample Collection and Preprocessing Protocol
The sample collection and preprocessing stages are critical in RNA extraction experiments, as they directly impact the quality of the final RNA yield. The rapid freezing of samples in liquid nitrogen is essential for preserving RNA integrity. For both animal and plant tissue samples, immediate immersion in liquid nitrogen is necessary after collection. The extreme low temperature of liquid nitrogen (-196°C) rapidly inactivates RNAse activity within the tissue, thus preventing RNA degradation. For example, promptly placing animal liver tissue into liquid nitrogen effectively maintains the original state of RNA.
Controlling the particle size during grinding is equally significant. After rapid freezing in liquid nitrogen, the animal tissue should be transferred to a pre-chilled mortar for grinding. Continuous addition of liquid nitrogen is crucial to ensure that the grinding process remains within a low-temperature environment until the particle size is ≤50μm. This particle size control facilitates the subsequent comprehensive lysis of cells by the Trizol reagent, allowing for efficient RNA release. In the case of plant tissue, which possesses a more complex cell wall structure, enhanced mechanical disruption is required. The liquid nitrogen-room temperature cycle freeze-thaw method (conducted for three cycles) is employed to rupture the cell wall prior to grinding.
The selection of RNase-free consumables is an important measure to prevent RNAse contamination. Throughout the experiment, it is imperative to utilize RNase-free centrifuge tubes, pipette tips, and glassware that has been treated by high-temperature baking (250°C for 4 hours). These RNase-free materials eliminate the risk of RNAse contamination, ensuring the accuracy of experimental results.
Different sample types require distinct handling procedures. Animal tissues should be treated as described above; plant tissues necessitate more stringent cell wall disruption; for cell samples, adherent cells should be digested using 0.25% trypsin, followed by centrifugation at 1000g for 5 minutes to collect the pellet, with Trizol added at a ratio of 1mL per 10cm² of culture area; for suspended cells, centrifugation should be performed prior to treatment at a ratio of 5 - 10×10⁶ cells/mL of Trizol.
A few operational prohibitions are essential to observe: first, if RNA extraction cannot be performed immediately after sample collection, the samples must be flash-frozen in liquid nitrogen and subsequently stored at -80°C to avoid RNA degradation due to prolonged exposure at room temperature; second, to minimize exposure to ambient air and prevent RNAse contamination, avoid frequently opening centrifuge tubes during the procedure; third, ensure pipette tips do not come into contact with items that may contaminate them with RNAse.
2. Key Steps in Phase Separation and Precipitation
The phase separation and precipitation steps of the Trizol method are pivotal for obtaining high-quality RNA and require strict control of parameters and operational techniques.
The ratio of chloroform added is a crucial factor during the phase separation process. After transferring the lysate to RNase-free centrifuge tubes, the precise addition of chloroform is fundamental; typically, this is 20% of the volume of Trizol used. This ratio ensures effective phase separation during subsequent centrifugation. For instance, if 1mL of Trizol is used, 200μL of chloroform should be added. Following the addition of chloroform, manually shake the mixture vigorously for 15 seconds to ensure thorough mixing, then let it sit for 2 - 3 minutes, during which the solution will gradually form a three-phase system.
The settings for centrifugation parameters significantly influence both the phase separation efficiency and the quality and yield of RNA. At 4°C and a speed of 12000g for 15 minutes, diverse components will separate effectively under the applied centrifugal force. The lower phenol-chloroform phase, having a greater density, contains hydrophobic proteins and DNA, while the middle denatured phase primarily consists of cellular debris, and the upper aqueous phase is enriched with RNA. Optimal centrifugation temperature and speed are essential to ensure that the phases are well-defined, facilitating subsequent processing.
The technique for aspirating the upper aqueous phase is critical for obtaining pure RNA. After centrifugation, a 200μL pipette should be used to slowly extract the supernatant along the wall of the tube, ensuring that the aspirating tip remains ≥2mm above the middle layer to avoid collecting impurities from this layer, which could contaminate the RNA and affect its purity and subsequent experimental results. For instance, inadvertent aspiration of material from the middle layer could introduce proteins and DNA into the RNA sample, leading to abnormal A260/A280 ratios and compromising RNA quality.
Among common pitfalls, contamination of the middle layer remains a prominent issue. Non-compliant practices, such as inserting the pipette too deeply or aspirating too rapidly, may result in the inclusion of cellular debris from the middle layer into the upper aqueous phase, thereby contaminating the RNA. Such contamination not only decreases RNA purity but may also interfere with subsequent experimental analyses, such as yielding anomalous bands during RNA electrophoresis or inaccurate data in quantification. Therefore, it is critical to adhere strictly to standardized protocols during the aspiration of the upper aqueous phase to ensure RNA quality.
Upon successful aspiration of the upper aqueous layer, the next step involves RNA precipitation. An equivalent volume of isopropanol (including 1μL of glycogen as a coprecipitant per mL of lysate) should be gently added to the supernatant, followed by gentle inversion of the tube 10 times to promote RNA precipitation through the action of isopropanol. After mixing, one may allow the mixture to sit for 10 minutes or overnight at -20°C to enhance precipitation effectiveness. Centrifugation at 12000g for 10 minutes at 4°C will yield a translucent gel-like pellet at the bottom of the tube, representing the desired RNA. When discarding the supernatant, it is crucial to maintain the tube in an inverted position for at least 30 seconds to ensure the complete removal of isopropanol, as residual isopropanol may impact subsequent RNA dissolution and experimental procedures.
3. RNA Dissolution and Storage Protocol
In the RNA dissolution phase, careful optimization of the volume of DEPC-treated water is necessary. The appropriate amount of DEPC water is critical for the complete dissolution of the RNA pellet. Generally, based on the amount of RNA precipitate, 30 - 50μL of DEPC water is typically added. If the volume is insufficient, the RNA may not fully dissolve, thereby affecting concentration determinations and subsequent usage; conversely, excessive addition may dilute the RNA solution too much, impairing experimental procedures. After adding the DEPC water, gentle pipetting or agitation should be employed to promote uniform dispersion and achieve optimal dissolution.
For RNA preservation, a strategy of aliquot freezing is essential for ensuring stability. The RNA should be dissolved in a buffer of 10 mM Tris - 1 mM EDTA (pH 8.0) and aliquoted at a volume of 50μL per tube, subsequently stored in siliconized freezing tubes at -80°C. This aliquot method prevents RNA damage due to repeated freeze-thaw cycles. Experimental data indicate that the integrity of RNA solutions experienced multiple -80°C/room temperature freeze-thaw cycles results in a marked decrease in RIN values, averaging a decline of 2.3±0.4 with each freeze-thaw cycle. Thus, aliquot freezing allows for the retrieval of only the necessary tube of RNA for each use, minimizing unnecessary freeze-thaw instances.
Prior to utilization, the frozen tube should be placed on ice for gradual thawing, ensuring that the structure and activity of the RNA remain unaffected. After use, any remaining samples should be promptly labeled and returned to the -80°C freezer for storage. Additionally, post-RNA dissolution, the concentration may be quantified using devices like Nanodrop to ensure it meets the requirements for subsequent experiments. Through the implementation of these measures, RNA can be effectively preserved, providing a stable and reliable sample for future molecular biology investigations.
Common Technical Issues and Solutions in RNA Extraction Process
I. Causes and Countermeasures for Insufficient RNA Yield
1.1 Inadequate Initial Sample Volume
Cause: A significantly reduced yield of RNA occurs when the cellular or tissue content in the initial sample falls below the threshold limits established by the reagent kit. A typical example is illustrated during the isolation of peripheral blood mononuclear cells, where an insufficient blood volume of less than 1 mL results in an inadequate number of leukocytes for RNA extraction.
Optimization Strategy: It is recommended to adopt a scaling approach based on sample type: for adherent cells, one could upgrade to larger culture plates (e.g., transitioning from a 6-well plate to a T25 flask); for suspended cells, increasing the culture volume can enhance cell density; tissue samples should be accurately weighed to meet the recommended weight threshold of the reagent kit (typically 50-100 mg), and if necessary, a tissue homogenization technique for pre-concentration can be employed.
1.2 RNase-Mediated Nucleic Acid Degradation
Mechanism: The hydrolysis of the β-glycosidic bonds in RNA molecules is highly sensitive to RNases, with potential sources of RNase contamination in the laboratory environment including human skin flakes (~10^4 U/mg) and aerosol particles. Notably, a case study indicated that if a denaturing buffer containing guanidine was not utilized during the grinding of skeletal muscle tissue, the RNA Integrity Number (RIN) could drop below 5.
Control Plan: Establish a four-tiered protective system: (1) Prepare solutions with 0.1% DEPC-treated water and autoclave them; (2) Wear powder-free gloves that have been baked at 160°C throughout the procedure; (3) Add 1 U/μL of recombinant RNase inhibitor (e.g., SUPERase·In™) to the lysis system; (4) Implement a low-temperature operational chain (using pre-chilled mortars at 4°C, performing lysis on ice, and centrifuging at 4°C).
1.3 Limitations of Extraction Reagent Efficacy
Technical Bottleneck: For plant samples with polysaccharide/polyphenol content exceeding 5% (such as pine needles or tubers), there is a notable reduction in binding efficiency with silica-based adsorption methods, which is linked to the competitive occupation of silica membrane binding sites by phenolic compounds.
Improvement Strategy: Employ a CTAB-β-mercaptoethanol composite lysis system in conjunction with LiCl precipitation. The specific workflow is as follows: (1) Add 2% CTAB lysis solution and incubate at 65°C for 30 minutes; (2) Use a 24:1 chloroform extraction to remove polysaccharides; (3) Add 1/4 volume of 10M LiCl for overnight precipitation at 4°C; (4) Wash with 75% ethanol and dissolve in DEPC-treated water.
1.4 Challenges in Processing Special Samples
Sample Characteristics: In adipose tissues (e.g., white adipose), triglyceride content exceeding 90% can hinder RNA-silica membrane binding, resulting in a 40-60% decrease in recovery rates.
Specialized Handling: Implement a three-step purification method: (1) Pre-cool PBS to wash away free lipids; (2) Add a five-fold volume of a 3:1 mixture of acetone-ethanol and de-fat at -20°C for 2 hours; (3) Centrifuge at 12,000×g for 10 minutes to collect the pellet and proceed with standard extraction protocols.
II. Optimization Solutions for Abnormal RNA Purity
2.1 Control of Protein Contamination
Contamination Source: Inadequate lysis volume (<5 times the sample volume) or inappropriate ultrasound parameters (>30% amplitude) can hinder the dissociation of nuclear membrane proteins from RNA, forming complexes.
Purification Technique: Utilize a gradient centrifugation method: (1) Perform an initial centrifugation at 12,000×g to remove cellular debris; (2) Follow with high-speed centrifugation at 100,000×g for 1 hour to precipitate nuclear proteins; (3) Employ HiBind® RNA adsorption columns for secondary purification.
2.2 Residual DNA Handling
Contamination Mechanism: When DNase I digestion is performed at temperatures exceeding 37°C or when Mg²⁺ concentrations are below 1 mM, the enzyme activity declines, leading to incomplete removal of genomic DNA.
Digest Optimization: Establish a standard digestion protocol: Add 10 μL of RNA sample to 2 U of DNase I and 1× reaction buffer (10 mM Tris-HCl, 2.5 mM MgCl₂, 0.5 mM CaCl₂), incubate at 25°C for 30 minutes, then add 2 mM EDTA to terminate the reaction.
2.3 Elimination of Organic Solvent Residues
Residue Detection: A spectrophotometric measure indicating an A230/A260 ratio greater than 0.5 suggests excessive phenolic contamination.
Purification Strategy: Employ a solid-phase extraction method: Mix the RNA solution with three volumes of binding buffer (5 M GuHCl, 20% ethanol) and pass through a column, washing sequentially with 70% ethanol and acetone (twice each), followed by drying at 65°C for 5 minutes before eluting.
III. Techniques for Maintaining RNA Integrity
3.1 Prevention of Mechanical Damage
Damage Threshold: When tissue homogenization occurs at speeds exceeding 25,000 rpm or for durations longer than 3 minutes, there is a significant decrease in the 28S/18S rRNA ratio.
Gentle Processing: Utilize low-temperature homogenization techniques: (1) Flash-freeze samples in liquid nitrogen to -196°C; (2) Grind in a pre-cooled ceramic mortar to a particle size of 50 μm; (3) Immediately transfer to RLT buffer containing β-mercaptoethanol.
3.2 Optimization of the Lysis System
Parameter Standards: A lysis system containing 4 M guanidine thiocyanate is recommended for mammalian cells (pH 6.4), while plant samples should utilize a CTAB system containing 2% PVP-40.
Condition Control: Accurately regulate the lysis temperature gradients: mammalian cells at 42°C for 5 minutes, Gram-positive bacteria at 65°C for 15 minutes, and lignified tissues at 70°C for 30 minutes (with vortexing for 5 seconds every 10 minutes).
3.3 Prevention of Freeze-Thaw Damage
Stability Study: Experimental data indicate that RNA solutions undergo an average RIN value decrease of 2.3±0.4 after three cycles of -80°C/room temperature freeze-thaw cycles.
Storage Scheme: The recommended approach is aliquot freezing: Dissolve RNA in a buffer of 10 mM Tris-1 mM EDTA (pH 8.0), aliquot into 50 μL per tube, and store in siliconized freezing tubes at -80°C. For use, place tubes on ice for thawing, and promptly refreeze any remaining samples after labeling.
More Issues in RNA Extraction refer to ”Troubleshooting Guide: RNA Extraction for Sequencing”.
RNA Quality Assessment and Downstream Applications
1. Purity Evaluation Using UV Spectrophotometry
Ultraviolet (UV) spectrophotometry is a widely utilized method for assessing RNA purity, primarily based on the key absorbance ratios of OD260/OD280 and OD260/OD230.
The OD260 measurement reflects the absorbance characteristics of nucleic acids, while OD280 is indicative of protein absorption. Theoretically, pure RNA exhibits an OD260/OD280 ratio of 2.0. In practical scenarios, a ratio within the range of 1.8 to 2.0 is considered acceptable. A ratio below 1.8 suggests potential protein contamination in the RNA sample, whereas a ratio exceeding 2.2 may indicate hydrolytic degradation of the RNA.
The OD260/OD230 ratio is specifically used to evaluate the levels of residual contaminants such as carbohydrates and salts, with a standard value expected to be greater than 2.0. A significantly low ratio suggests the presence of residual reagents such as guanidine isothiocyanate, β-mercaptoethanol, or ethanol. For instance, an OD260/OD230 ratio of 1.5, which is markedly below the standard value, indicates substantial residual impurities. In such cases, strategies such as repeating the precipitation process or increasing the number of ethanol washes can be employed for purification.
For example, if a certain sample has an OD260/OD280 ratio of 1.6, which is below the normal range, it suggests possible protein contamination. Additionally, an OD260/OD230 ratio of 1.2, significantly below the standard, indicates the presence of excessive organic solvent residues. To address this, the sample may first undergo additional ethanol washes to remove some impurities, followed by repeated precipitation for enhanced RNA purity, ensuring compliance with subsequent experimental requirements.
2. Integrity Assessment via Gel Electrophoresis and Bioanalyzer
In evaluating RNA integrity, both agarose gel electrophoresis and the Agilent 2100 Bioanalyzer are common methods, each offering unique features that provide critical information regarding RNA quality.
Agarose gel electrophoresis is a classic detection method. Upon electrophoretic separation, intact RNA from eukaryotic sources presents three characteristic bands corresponding to 5S, 18S, and 28S rRNA. By observing these bands, researchers can preliminarily assess RNA integrity. Typically, a clear presence of the 28S band at approximately twice the intensity of the 18S band indicates good RNA integrity, as intact RNA maintains structural stability during electrophoresis, resulting in a distinct band distribution. Conversely, the appearance of diffuse or blurred bands, or an abnormal intensity ratio, such as a significantly weaker 28S band compared to the 18S band, suggests notable RNA degradation. However, the interpretation of results from agarose gel electrophoresis is somewhat subjective, and it may be challenging to accurately assess mild degradation.
The Agilent 2100 Bioanalyzer, on the other hand, offers a more precise quantitative assessment. Utilizing microfluidic chip electrophoresis technology, it provides detailed analysis of RNA and outputs a RNA Integrity Number (RIN). RIN values range from 1 to 10, with values ≥7 considered indicative of high-quality RNA, suitable for a variety of experiments that require stringent RNA quality. RIN values within the range of 6 to 7 indicate partial degradation but may still permit usage in certain assays. When the RIN value is <6, the RNA is deemed inadequate in quality and may not fulfill the requirements for subsequent applications. This quantifiable assessment provides a more objective and accurate reference for researchers.
In practical applications, thresholds for determining sample degradation differ between methods: agarose gel electrophoresis relies on the morphology and intensity ratios of the bands, while the Agilent 2100 Bioanalyzer uses a RIN threshold of 6 as a crucial cutoff point. Researchers can choose suitable detection methods based on specific experimental needs and sample conditions to effectively evaluate RNA integrity, thereby ensuring the successful progression of subsequent experiments.
3. Verification of Compatibility for Downstream Experiments
Different downstream experiments impose varying requirements on the quality and characteristics of RNA samples.
In the case of quantitative real-time polymerase chain reaction (qPCR), it is generally required that the RNA's OD260/OD280 ratio falls within the range of 1.8 to 2.0. This criterion ensures sample purity and minimizes interference from contaminants such as proteins. Additionally, a RIN value of ≥7 is preferred to confirm RNA integrity, providing a reliable foundation for accurate quantitative assessment of gene expression. Regarding fragment distribution, intact mRNA segments are critical to avoid significant degradation, which could lead to abnormal primer binding and subsequently affect amplification efficiency and the accuracy of results.
For RNA sequencing (RNA-seq), the quality requirements are even more stringent. An OD260/OD280 ratio close to 2.0 and an OD260/OD230 ratio exceeding 2.0 are essential to ensure high-purity samples. A RIN value of ≥8 is necessary to guarantee high RNA integrity, facilitating the construction of quality libraries for accurate whole transcriptome coverage. Furthermore, the distribution of RNA fragments should be uniform, avoiding the prevalence of large segment deletions or small fragment enrichments that could compromise sequencing data quality and subsequent analyses.
Only by meeting these stringent criteria can one ensure the smooth execution of downstream experiments and obtain reliable experimental outcomes.
Specialized Sample Extraction Techniques
1. RNA Extraction from Plant Samples Containing Polysaccharides and Polyphenols
Extracting RNA from plant samples rich in polysaccharides and polyphenols presents significant challenges, as these compounds can co-precipitate with RNA, adversely affecting its purity and yield. Effective processing techniques for such samples include the modified CTAB buffer method and LiCl gradient precipitation.
Taking lignified tissues as an example, the process begins with the application of the modified CTAB buffer method. An appropriate amount of lignified tissue is placed into a mortar, ground thoroughly in liquid nitrogen to create a powdered form that disrupts the cell walls. The powder is then quickly transferred into a centrifuge tube containing CTAB lysis buffer and incubated at 65°C for 30 minutes, gently inverting the tube to ensure complete cell lysis and RNA release. The CTAB buffer effectively removes polysaccharides and inhibits polyphenol oxidation.
Following this step, LiCl gradient precipitation is conducted. After the incubation, the sample is centrifuged at 12,000g for 15 minutes at 4°C, and the supernatant is transferred to a new centrifuge tube. To this, 1/4 volume of 10M LiCl solution is added, and it is allowed to precipitate at 4°C overnight. LiCl selectively precipitates RNA while removing some impurities. The next day, the RNA pellet is washed twice with 75% ethanol to eliminate residual LiCl and other contaminants. Finally, the RNA pellet is dissolved in DEPC-treated water, yielding high-quality RNA samples.
Through these two steps, the challenges associated with RNA extraction from plant samples containing polysaccharides and polyphenols can be effectively addressed, providing a reliable sample basis for subsequent experiments.
2. Cell Wall Disruption Strategies for Microbial Samples
In the extraction of RNA from microbial samples, particularly from Gram-positive bacteria and yeast, the structural composition of their cell walls presents a key challenge for efficient cell lysis. Commonly employed lysozyme treatments and mechanical disruption methods each have their advantages and limitations; therefore, a combined approach often yields optimal results.
Lysozyme treatment is effective for Gram-positive bacteria, specifically hydrolyzing the peptidoglycan in the cell wall, which weakens the structural integrity and facilitates RNA release. However, for yeast samples with more complex cell wall structures, lysozyme alone may prove insufficient, and prolonged exposure could compromise RNA quality.
Mechanical disruption methods, such as sonication or grinding, apply physical force to rupture the cell walls. For yeast samples, mechanical disruption effectively breaks open the tough cell wall, though it may generate localized heat, which can cause RNA degradation.
A combined strategy may involve initially treating Gram-positive bacteria with lysozyme, followed by mild mechanical disruption (e.g., gentle sonication) to minimize RNA damage. For yeast samples, a moderate mechanical disruption can first be employed to open the outer cell wall structure, which is then followed by lysozyme treatment to enhance lysis efficiency. Careful control of processing conditions is crucial to prevent RNA degradation and ensure the extraction of high-quality RNA.
3. Optimization of RNA Extraction from Clinical FFPE Samples
Clinical formalin-fixed paraffin-embedded (FFPE) samples offer significant value in disease research; however, the fixation and embedding processes typically result in poor RNA quality and extraction challenges. To enhance the success rate of RNA extraction from these low-quality samples, several optimization measures can be implemented.
The first critical step is deparaffinization. Thin sections of the FFPE samples should be immersed in xylene to dissolve the paraffin, thereby releasing the tissue. The deparaffinization time should be adjusted based on the thickness of the sections and the amount of paraffin, generally ranging from 15 to 30 minutes, with regular changes of xylene to ensure effective removal. Following deparaffinization, a gradient ethanol treatment is performed to hydrate the tissue, bringing it closer to a fresh state and preparing it for subsequent digestion and extraction.
Controlling the duration of proteinase K digestion is also essential. Proteinase K breaks down proteins, releasing RNA that is bound to these proteins. However, prolonged digestion can lead to excessive degradation of RNA, while insufficient digestion may leave proteins intact, hindering RNA release. Typically, incubating at 55-60°C for 2-4 hours is advised, with the specific duration further optimized based on sample type and the activity of proteinase K.
For damaged RNA, RNA repair techniques can be employed to recover integrity. Several commercial kits are available that provide specific RNA repair enzymes capable of mending broken RNA strands and enhancing RNA integrity. During the extraction process, the recommended amount of RNA repair enzyme should be added according to the kit instructions and incubated under specified conditions to effectively improve RNA quality.
Moreover, throughout the extraction process, it is imperative to use RNase-free reagents and consumables to prevent RNA contamination. Additionally, strict control of experimental temperatures and time should be maintained to minimize RNA degradation. By implementing these optimization strategies, the quality of RNA extracted from clinical FFPE samples can be significantly enhanced, providing reliable sample support for subsequent molecular biology experiments.






