Single-Cell RNA-Seq
Overview
Features
Project Workflow
Bioinformatics Analysis Pipeline
Sample Requirements
Demo Results
Cases & FAQ
Resources
Inquiry
Single-cell RNA sequencing (scRNA-seq) is a next-generation sequencing (NGS)-based method to amplify and sequence the whole transcriptome of a single cell. It is becoming a powerful tool and has been applied to research related to stem cell differentiation, embryogenesis, whole tissue analysis, and even tumors.
Overview
Single-cell RNA-seq is a method that enables simple and comprehensive access to the transcriptome of thousands of single cells. The principle is to separate multicellular organisms into single cells, and then the RNA of single cells is efficiently amplified using mature whole genome amplification (WGA) kits before being subjected to high-throughput sequencing. Single-cell RNA-seq allows a comparison of the transcriptome of individual cells and reveals the similarities and differences of the transcriptome in cell populations. scRNA-seq involves single-cell isolation, cell lysis, reverse transcription of RNA to cDNA, optional polyA RNA selection, followed by DNA sequencing. Unlike the standard bulk RNA sequencing, scRNA-seq provides unparalleled details of different cells in a sample, allowing researchers to move from average data for large numbers of tissues to individualized cellular expression data.
From basic research to clinical applications, there are many applications for scRNA-seq. It provides transcriptome information about single cells, enabling researchers to analyze the unique gene expression patterns of each cell, understand cellular heterogeneity, and explore how different cells promote disease progression and clinical response. Currently, this technique has been changing our understanding of basic biological processes, such as development, immunity or cell-related pathology.
Features
| Any Species |
Single-Cell Insights |
High Resolution |
Multiple Applications |
| This method can be applied to any species, from microorganisms to humans. |
Quantitative analysis down to single-cell levels for input samples. |
Discovery of more cellular differences based on high resolution analysis. |
Understanding complex tissues, tumor heterogeneity and clonal evolution. |
Project Workflow

1. Sample Preparation
Isolation of viable, single cells from a given sample
2. Library Preparation
Total RNA or poly-A RNA; Smart-seq2

3. Sequencing
Illumina HiSeq; PE50/75/100/150; >10G clean data

4. Data Analysis
Provide customized bioinformatics analyses and services for users.
Bioinformatics Analysis Pipeline
Comprehensive analysis:
- Advanced cell type characterization
- Refinement of destination cell populations through reclustering
- Gene set variation analysis (GSVA)
- Integrated differential expression analysis (iDEA)
- Evolution of cellular population states
- Proposed timing analysis
- Rate analysis for cellular transitions
- Trajectory exploration
- Investigation of cell-cell interactions
- In-depth cell cycle analysis
- Assessment of regulatory factors
- Detection of single-cell mutations
- Comprehensive single-cell copy number variation (CNV) analysis
- Identification of subpopulation Outcomes
Integration analysis
Sample Requirements
- Live cells: 1-1000 intact cells from all vertebrate species. 96-well plates of live cells in suspension are recommended.
- RNA: Purified RNA sample quantity ≥ 100 pg, 1.8 ≤ OD260/280 ≤ 2.0.
- Please make sure that the RNA is not degraded.
Deliverable: FastQ, raw data, coverage summary, QC report, experiment results, custom bioinformatics analysis.
Demo Results
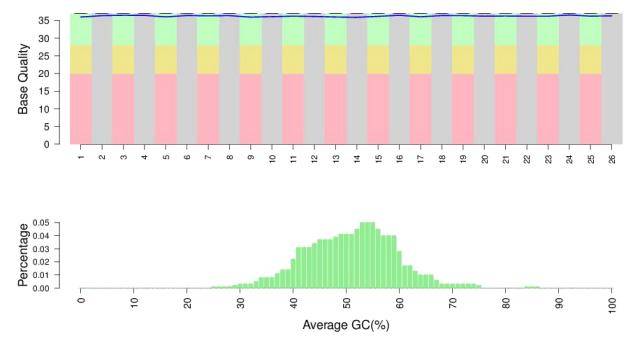 Data quality control
Data quality control
 Cell filtration and statistics
Cell filtration and statistics
 Dimensionality reduction and clustering
Dimensionality reduction and clustering
 Identification of cell subpopulations
Identification of cell subpopulations
 Differentially expressed gene analysis
Differentially expressed gene analysis
 KEGG pathway functional analysis
KEGG pathway functional analysis
 Pseudotime trajectory analysis
Pseudotime trajectory analysis
Case Studies
-
Single-Cell Transcriptome Sequencing Reveals Key Fibrotic Drivers
-
This case delves into an examination of the cellular composition within the bone marrow stroma of both mice and humans. The study leveraged single-cell RNA sequencing, specifically focused on purified non-hematopoietic bone marrow cells, to pinpoint fibrotic driver cells and elucidate potential mechanisms in Myeloproliferative Neoplasms (MPNs).
The study uncovered two distinct populations of mesenchymal stromal cells (MSCs) denoted as MSC-1 and MSC-2, which emerged as the central instigators of fibrosis in bone marrow MSCs. Further investigation also unveiled two separate subpopulations of mesenchymal stromal cells with pro-fibrotic characteristics. Notably, the MSCs exhibited a dynamic shift in function during different stages of MPNs. They initiated differentiation prior to fibrosis and adopted pro-fibrotic as well as inflammatory traits during the fibrotic stage.
Moreover, the research identified the expression of the protein complex S100A8/S100A9 in MSCs as a marker for the disease's progression toward the fibrotic stage, observed in mouse models, patient stroma, and plasma. Significantly, Tasquinimod, a small molecule capable of inhibiting S100A8/S100A9 signaling, showed promising results in ameliorating the MPN phenotype and fibrosis in the JAK2V617F mutant mouse model. This discovery points towards S100A8/S100A9 as a compelling therapeutic target for MPN treatment, potentially offering valuable insights for future myeloma therapy
 Single-cell transcriptome sequencing reveals key fibrotic drivers. (Leimkühler et al., 2021)
Single-cell transcriptome sequencing reveals key fibrotic drivers. (Leimkühler et al., 2021)
-
Unveiling Physiological and Developmental Insights in Rice Seedlings and Roots by Single-Cell Transcriptome Analysis
-
In this study, the authors harnessed the power of single-cell RNA sequencing to dissect the intricacies of rice seedling leaves and root systems. By conducting a comprehensive analysis of 237,431 individual cells, they shed light on the physiological and developmental mechanisms at play in these vital plant organs. The investigation encompassed various growth conditions, ranging from nutrient-rich environments to the imposition of diverse abiotic stresses.
The research methodology entailed single-cell RNA sequencing on both stems and roots of rice seedlings grown under two distinct conditions: in a nutrient-rich Kimura B solution and under a variety of abiotic stresses. This extensive dataset provided a foundation for characterizing the transcriptome of each individual cell.
 Quality control experiments for the application of scRNA-seq to rice seedlings. (Wang et al., 2021)
Quality control experiments for the application of scRNA-seq to rice seedlings. (Wang et al., 2021)
The authors unveiled 15 distinct cell types within rice leaves and 9 cell types within the root system. Remarkably, they identified common transcriptome features shared between leaves and roots, specifically within the same tissue layer, offering insight into the genetic commonalities that underpin these crucial plant structures.
Furthermore, the impact of abiotic stressors on gene expression was examined with a cell type-specific focus. It was observed that different stresses, while eliciting unique responses, tended to regulate the transcription of a largely conserved set of genes within a particular cell type. This finding highlighted the adaptive flexibility of rice seedlings to environmental challenges.
Additionally, the study delved into the shifts in cell population proportions in response to abiotic stresses. By reconstructing developmental trajectories from single-cell data, the authors elucidated the molecular mechanisms governing these alterations in cell populations. This approach yielded valuable insights into the dynamic responses of rice seedlings to external stressors.
FAQ
-
How to decide between bulk transcriptome and single-cell transcriptome sequencing?
-
Bulk RNA-seq captures the collective transcription of all mRNAs within a biological tissue sample at a specific point in time, serving as a valuable indicator of the sample's overall state. However, conventional transcriptome sequencing yields an average expression profile across all cells, failing to portray the individual states of each cell or specific cell subsets within the sample. In contrast, single-cell transcriptome sequencing offers a comprehensive exploration of the transcriptional landscape within a single cell or a defined group of cells. This approach delivers a more granular and precise assessment of the tissue's condition.
-
When preparing single-cell suspensions, what type of dissociation enzyme is appropriate to choose for different tissues?
-
When creating single-cell suspensions, the selection of an appropriate dissociation enzyme depends on the specific tissue type in question. It is crucial to pick the enzyme or enzyme combination that aligns with the tissue's characteristics. The hierarchy of enzyme effectiveness for dissociation is as follows: trypsin > papain > elastase > hyaluronidase > collagenase > neutral protease. Some tissues may require specialized dissociation kits readily available in the market. For more precise guidance tailored to specific requirements, we recommend consulting our Sample Submission Guidelines or reaching out to our technical support team.
-
If the single-cell suspension resulting from dissociation meets all criteria except for cell activity, which falls below 85%, can it still be processed using the machine?
-
When the cell activity hovers around 85%, it is acceptable for the customer to proceed with machine processing. However, if the cell activity is exceptionally low (e.g., approximately 60% or even lower), it is advisable not to run the experiment directly on the machine. In such cases, the recommended approach is to eliminate the non-viable cells, ensuring that the remaining cell population meets the required activity standard, and then proceed with machine-based experimentation.
-
When doing parallel experiments, it is found that the samples from the same source, the results of downscaling clustering and sub clustering are very different, how to avoid it?
-
- Batch Effect Mitigation: Batch effects are a common source of variability in high-throughput experiments. Employ strategies such as batch correction methods during data analysis to minimize the impact of these technical variations. This will help ensure more consistent results between experiments.
- Normalization Techniques: Apply appropriate data normalization techniques to adjust for differences in sequencing depth, library preparation, or other technical factors. Normalization helps ensure that the data from different experiments are on a comparable scale.
- Quality Control: Implement rigorous quality control measures to identify and address issues like low-quality cells, outliers, or technical artifacts that can introduce variability in the results.
- Replicate Analysis: Whenever possible, increase the number of replicates for each experiment to enhance statistical power and confidence in the results. Consistency is more likely to be achieved with a larger sample size.
- Consistent Protocols: Ensure that sample collection, processing, and library preparation protocols are standardized and consistent across experiments to minimize sources of variation.
- Parameter Tuning: Fine-tune analysis parameters, such as clustering algorithms and dimensionality reduction techniques, to suit the specific characteristics of your data.
-
How to approach single-cell sequencing when the cell count is insufficient?
-
Single-cell sequencing demands optimal cell activity and a sufficient starting cell volume, typically requiring a minimum of 10^5 cells. When facing a scarcity of cells, consider the following strategies to enhance the cell count:
- Sample Mixing: In cases where tissue samples are too small, consider combining multiple samples to create a single-cell suspension. This ensures the dissociation of a more substantial number of cells.
- Flow Enrichment: Increase cell numbers by employing flow enrichment techniques, where multiple samples are pooled for single-cell sequencing, thus boosting the overall cell count.
- Whole Tissue Dissociation: When dealing with a specific cell type that constitutes a small proportion of the total cells, consider dissociating the entire tissue. This approach increases the capture of the target cell population to over 10,000 cells, as long as the target cell type constitutes at least 1% of the total. Subsequently, the target cell population data can be extracted for focused subgroup analysis and differential analysis, facilitating specific research on the target cell group.
-
What is the optimal data volume for single-cell sequencing?
-
For single-cell sequencing, targeting a sequencing data size of approximately 50,000 read pairs per cell is recommended, equating to approximately 100 gigabytes of clean data per sample. When aiming for 100 gigabytes of sequencing data, the number of genes that can be reliably detected tends to stabilize. Additionally, there is a modest increase in the detection of new genes as the amount of sequencing data is further increased.
-
Is utilizing the Illumina platform for sequencing the libraries created during library construction mandatory?
References:
- Leimkühler, Nils B., et al. "Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis." Cell Stem Cell 28.4 (2021): 637-652.
- Wang, Yu, et al. "Single-cell transcriptome atlas of the leaf and root of rice seedlings." Journal of Genetics and Genomics 48.10 (2021): 881-898.
* For Research Use Only. Not for use in diagnostic procedures.












